SCOPe 2.08 help
Overview
Structural Classification of Proteins — extended (SCOPe) is a database of protein structural relationships that extends the SCOP database. SCOP is a (mostly) manually curated ordering of domains from the majority of proteins of known structure in a hierarchy according to structural and evolutionary relationships. Development of SCOP (version 1) concluded with SCOP 1.75, released in June 2009. SCOPe extends the SCOP database, using a combination of manual curation and rigorously validated automated methods to classify many newer PDB structures. Our goal in implementing automation is to extend SCOPe to include structures that can be classified in the SCOP hierarchy, without sacrificing the reliability of the database that was developed through years of expert curation.
SCOPe also incorporates and updates the Astral compendium. Astral provides several databases and tools to aid in the analysis of the protein structures classified in SCOP, particularly through the use of their sequences. Here is a link to more info about Astral. New releases of Astral are derived from SCOPe.
We have rigorously benchmarked our automated methods to ensure that they are as accurate as manual curation, though there are many proteins to which our methods cannot be applied. SCOPe also corrects some errors in SCOP. SCOPe aims to be backward compatible with SCOP, providing the same parseable files and a history of changes between all stable SCOP and SCOPe releases (available on the Stats & History page).
The SCOPe website provides integrated access to data found in all releases of the SCOP and Astral databases that feature stable identifiers.
The SCOP hierarchy
- Applies to: SCOP version 1.55 through current release
- Reference: 5

For backward compatibility, SCOPe entries are arranged in the same hierarchy as SCOP (version 1) entries.
By analogy with taxonomy, SCOP was created as a hierarchy of several levels where the fundamental unit of classification is a domain in the experimentally determined protein structure. Starting at the bottom, the hierarchy of SCOP domains comprises the following levels:
- Species representing a distinct protein sequence and its naturally occurring or artificially created variants.
- Protein grouping together similar sequences of essentially the same functions that either originate from different biological species or represent different isoforms within the same species
- Family containing proteins with similar sequences but typically distinct functions
- Superfamily bridging together protein families with common functional and structural features inferred to be from a common evolutionary ancestor.
- Folds grouping structurally similar superfamilies.
- Classes based mainly on secondary structure content and organization.
Only the first seven classes are true classes. Because traditionally the aim of SCOP has been to classify every residue in the PDB, the remaining classes were maintained as placeholders to keep track of portions of a PDB structure that were not appropriate to include in SCOP. The SCOPe automated classification methods do not currently add entries to classes other than the first seven.
The multi-domain class is a special class. It contains chains with multiple domains that haven't been observed in a different context.
Changes to SCOP(e) design and size
- Applies to: SCOP version 1.55 through current release
- References: 1-5,8
The figure below shows the number of structures in the PDB and
SCOP(e) at the time of
each SCOP(e) release. Extended
horizontal lines start at the freeze date for
each SCOP(e) release and show the
number of PDB entries available on that date. (The "freeze date"
is the last date for PDB entries to be released and still classified
in a given SCOP(e) release. Prior to
SCOP 1.73, all protein structures available on
the freeze date were manually classified.)


The figure below, adapted from
[3],
illustrates how SCOP(e)
has changed over the years. The length of the vertical line for
each release is proportional to the number of entries classified. The angle
of the blue baseline between releases reflects the degree of
divergence from comprehensive and fully manually curated
releases. The angle is based on the percentage of PDB entries on the
freeze date that were not classified in the release, and the extent
to which entries were manually classified.
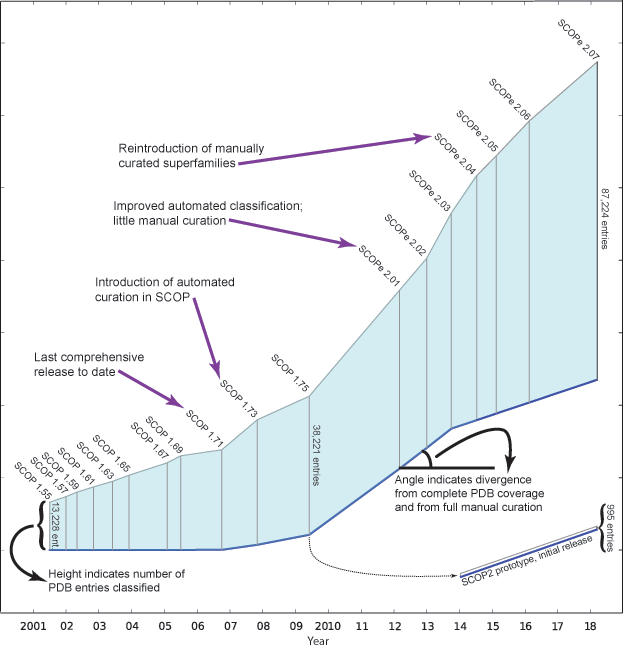
Note that since releases beyond SCOP 1.71 are not comprehensive, not all structurally characterized protein families and folds from the PDB are classified in these releases. Therefore, we caution against using later releases to (for example) analyze the rate at which new folds are being discovered.
What is the relationship between SCOP, SCOPe, and SCOP2?
- Applies to: SCOPe version 2.01 through current release
- References: 1-5
SCOP (version 1) is a database that originated in 1994 for protein structure classification [PubMed]. The last version of this database, 1.75, was released in June 2009 and is still available both in Cambridge UK and on the SCOPe website. SCOP 1.75 classifies 38,221 PDB entries.
SCOPe continues the development and maintenance of the classic SCOP hierarchy and may be seen as a continuation of the SCOP database, with ongoing updates and new classifications [PubMed]. SCOPe is completely backward compatible with SCOP (version 1), including all the same parseable files that we previously released separately in SCOP and Astral releases. As of March 2018, SCOPe 2.07-stable classifies 87,224 PDB entries.
SCOP2 represents a marked change from the SCOP classification structure and is currently available as a prototype with only a small number of classified families. SCOP2 replaces the tree-like SCOP hierarchy with a directed acyclic graph, allowing curators to represent structural and evolutionary relationships between proteins in more precise detail. There is no division of proteins into domains with fixed boundaries; instead, node-specific bounaries define the protein regions for each particular relationship [PubMed]. As of March 2018, the SCOP2 prototype classifies 995 PDB entries.
Stable identifiers
- Applies to: SCOP version 1.55 through current release
- Reference: 8
The SCOPe database continues to support the same style of stable identifiers in use since SCOP 1.55. Identifiers are provided as an unambiguous way to link to each a SCOP or SCOPe entry and are stable across releases. See the sections on citing and linking to SCOPe and SCOP using stable identifiers.
-
sccs. SCOP(e) concise classification string. This is a dot notation used to concisely describe a SCOP(e) class, fold, superfamily, and family. For example, a.39.1.1 references the "Calbindin D9K" family, where "a" represents the class, "39" represents the fold, "1" represents the superfamily, and the last "1" represents the family.
-
sunid. SCOP(e) unique identifier. This is simply a number that may be used to reference any entry in the SCOP(e) hierarchy, from root to leaves (Fold, Superfamily, Family, etc.).
-
sid. Stable domain identifier. A 7-character sid consists of "d" followed by the 4-character PDB ID of the file of origin, the PDB chain ID ('_' if none, '.' if multiple as is the case in genetic domains), and a single character (usually an integer) if needed to specify the domain uniquely ('_' if not). Sids are currently all lower case, even when the chain letter is upper case. Example sids include d4akea1, d9hvpa_, and d1cph.1.
Both sunids and sccs identifiers are expected to remain stable across releases, except in cases where the classification changes substantially. For example, when nodes in the hierarchy (e.g. Superfamilies) are merged or split due to new evidence of evolutionary relationships, corresponding identifiers become obsolete and new sunids are introduced. If a domain is split, or the boundaries change substantially, new sid(s) and sunid(s) are assigned.
A history of changes between all consecutive SCOP(e) releases is available under the Stats & History tab, and a history of changes to each individual entry is shown at the bottom of the page describing that entry.
Astral documentation
- Applies to: SCOP version 1.55 through current release
- References: 11-13
The primary sources of Astral documention are the three references cited above and listed on the Help > References page. The Astral compendium is a collection of software and databases, partially derived from SCOP(e), that aid research into protein structure and evolution. Astral provides sequences and coordinate files for all SCOP(e) domains, as well as sequences for all PDB chains that are classified in SCOP(e). Chemically modified amino acids are translated back to the original sequence, and sequences are curated to eliminate errors resulting from the automated parsing of PDB files. Because the majority of sequences in the PDB are very similar to others, Astral provides representative subsets of proteins that span the set of classified protein structures or domains while alleviating bias toward well-studied proteins. The highest quality representative in each subset is chosen using AEROSPACI scores, which provide a numeric estimate of the quality and precision of crystallographically determined structures.
The figure below, taken from the
2004 NAR paper, gives a brief overview
of how Astral is created. New releases
of Astral are derived from SCOPe domain definitions.

Data flow in Astral: Primary data sources are shown in green. Primary Astral databases are shown in light yellow. Less commonly used resources are shown in darker yellow. Resources added more recently are outlined in light blue/grey. Using the RAF maps, four complete sequence sets are created for every domain in the first seven classes of the SCOP(e) database. Two sets (the genetic domain sets) include the genetic domain sequences described in the 2002 NAR paper, and the other two (the original-style sequence sets) use the prior method of splitting each multi-chain domain into multiple sequences. For each of these methodologies, one complete sequence set is derived from sequences in the PDB ATOM records, and another from sequences in the SEQRES records. The SEQRES sets (for both genetic domain and original-style methods) are used to derive representative subsets. Each set is fully compared against itself using BLAST, and subsets are created using three similarity criteria and various thresholds. Representatives are chosen according to AEROSPACI scores. PDB chain sequence sets are derived from the SEQRES records of every PDB chain in SCOP(e); selected subsets are created at 90-100% ID thresholds. PDB-style files are derived from the RAF maps and SCOP(e) domain definitions. At each new release of Astral, all non-redundant sequences from each SCOP(e) family and superfamily are aligned using MAFFT. A hidden Markov model (HMM) is created from the multiple sequence alignment for each family and superfamily using HMMER. These HMMs, and BLAST, are used to predict domains in the sequences of newly released PDB entries on a weekly basis. HMMs from the Pfam-A database are also used to predict domains in regions of the sequences not identified by HMMs derived from Astral. Unassigned regions of at least 20 consecutive residues are also predicted to be potential domains.
Documentation describing more recent updates (since the 2004 paper) can be found in the release notes, which are linked from the Downloads > Astral Sequences & Subsets page.
Searching SCOP and SCOPe
- Applies to: SCOP version 1.55 through current release
- Reference: 8
The website search bar at the top of each page supports keyword search, as well as searches by a number of different SCOP(e) identifiers.
View the Search examples page for further details on the syntax, as well as many examples.
Variant Search
- Applies to: SCOPe version 2.08 through current release
- Reference: 2
The variant viewer displays a summary of the predicted impact, an interactive viewer showing the structural context of the variant, and relevant evolutionary context from the SCOPe hierarchy, including members of the same family or superfamily as the impacted protein domain. In the example shown below, the user searched for a missense variant in chromosome 2, which affects the coding sequence of the ZAP-70 protein. The variant viewer displays the most relevant human ZAP-70 structure classified in SCOPe. Note that in this structure, the amino acid residue affected by the variant is located in a structurally uncharacterized loop in the protein, so the nearest residues in the structure are highlighted. Several additional ZAP-70 structures are also shown, allowing users to visualize the impact of the variant in different structural contexts.

Domain visualization
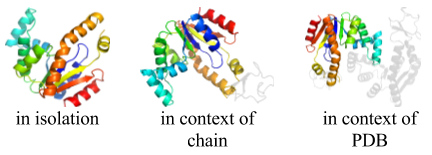
- Applies to: SCOP version 1.55 through current release
- Reference: 1
Thumbnails were generated for each domain using PyMOL, based on a viewing angle for each protein calculated using OVOP (Sverud O, MacCallum RM. 2003. Towards optimal views of proteins. Bioinformatics 19(7): 882-888 [PubMed]).
On the SCOPe website page showing information about each domain, thumbnails are displayed showing the domain in isolation, in the context of its chain, and in the context of its PDB structure. Links to other domains in the same chain, and in the same PDB structure, are below the corresponding thumbnail. To preview thumbnails from these other domains and get tool tip text with a short description, mouse over the links to the other domains. The figure to the right shows the thumbnails generated for the domain d4akea1.
Domain pages also include a JavaScript-based viewer that allows users to view and rotate domains in 3D without installing additional software. The 3D visualization domain visualization tool was built using JSmol, a Javascript-based viewer created by the Jmol project.
How to link to SCOP and SCOPe nodes
- Applies to: SCOP version 1.55 through current release
- References: 1,8
Because the SCOPe website is based on different technology from the older SCOP websites, old search paths (e.g., those using the search.cgi engine) are deprecated. URLs in the older style will be redirected approprately, so all old links to the Berkeley SCOP mirror should still work, including links to retrieve data that used to be on the Astral website (e.g., getseqs.cgi, spaci.cgi). We also support old-style links to all versions of SCOP with stable identifiers; for example, http://scop.berkeley.edu/scop/ links to SCOP 1.75, the last release of SCOP (version 1), while http://scop.berkeley.edu/scop-1.67/ links to SCOP version 1.67.
When making new links to SCOP or SCOPe data on this website, please use the new link formats below:
-
To link to a node the current release, use any
stable identifier (sunid,
sid or sccs).
The link formats are:
http://scop.berkeley.edu/sunid=xxxxx http://scop.berkeley.edu/sid=xxxxx http://scop.berkeley.edu/sccs=xxxxx
Linking by sunid is preferred because it is faster (sunid is a number, e.g. 46456; sids and sccs are both alphanumeric). Note that if the identifier you are linking to has become obsolete in the current release, the last release in which the identifier appears is shown, along with an explanation. -
To link to a node in a particular SCOP
or SCOPe
release, use the version number and sunid. The link format is:
http://scop.berkeley.edu/sunid=xxxxx&ver=yyyy
(versions are alphanumeric; e.g., 1.55 or 2.01).
You can also use this style of linking to link to nodes by sid or by sccs. -
To link to the root of a particular SCOP
or SCOPe
release, use the version number. The link format is:
http://scop.berkeley.edu/ver=yyyy
(versions are alphanumeric; e.g., 1.55 or 2.01). -
To link to information we have on a PDB
file, use its PDB code. The link
format is:
http://scop.berkeley.edu/pdb/code=xxxx
We plan for this link format to remain stable for all future versions of SCOPe unless there is a major change to the classification, even if we make changes to our underlying website technology.
How to cite SCOP and SCOPe entries
- Applies to: SCOP version 1.55 through current release
- References: 1,8
In the interest of scientific reproducibility, when you publish a manuscript or give a talk about data you obtained from SCOP or SCOPe, it's crucial to unambiguously refer to the data you used. Using these citation conventions will provide all the information needed for other researchers to accurately download the same data that you are working with. For related help, see sections on backward compatibility with SCOP, stable releases and updates, and stable identifiers.
When citing a particular SCOPe or SCOP entry, we recommend the following conventions, depending on the level of the entry cited:
- Class: Use the complete SCOP(e)
version number, full name of the class, and
sunid. Examples:
- SCOPe 2.03-stable Class a: All alpha proteins [46456]
- SCOP 1.67 Class c: Alpha and beta proteins (a/b) [51349]
- Fold, Superfamily, or Family: Use the complete SCOP(e)
version number, sccs,
full name, and sunid. Examples:
- SCOPe 2.03-2013-12-12 Fold b.1: Immunoglobulin-like beta-sandwich [48725]
- SCOPe 2.03-stable Family c.2.1.1: Alcohol dehydrogenase-like, C-terminal domain [51736]
-
Protein or Species: Use the complete SCOP(e)
version number, full
name, sunid, and sccs. Examples:
- SCOPe 2.03-stable Protein Adenylate kinase [52554] (sccs c.37.1.1)
- SCOP 1.75 Species: Baker's yeast (Saccharomyces cerevisiae) [TaxId: 4932] [56639] (sccs e.5.1.1)
-
Domain. Use the complete SCOP(e)
version number, sid, sunid, and sccs. Examples:
- SCOPe 2.03-stable d1p0ca2 [87639] (sccs c.2.1.1)
- SCOPe 2.03-2013-12-12 d4cadj2 [229759] (sccs b.1.1.0)
Publications using SCOP, Astral or SCOPe data should also cite the appropriate references given on the Help > References page rather than just referring to the website.
Parseable files
- Applies to: SCOP version 1.55 through current release
- References: 2,8
Parseable files have been provided for each SCOP(e) release, available from the Downloads > Parseable Files & Software page. Each of these files has a header, starting with the '#' character, which includes release, version, and copyright information.
The list below shows the six parseable formats for SCOP(e) data. For information on downloading and parsing Astral subsets and RAF sequence maps, see the Astral help.
-
dir.des.scop(e).txt. - Descriptions for each node in the SCOP(e) hierarchy.
46457 cf a.1 - Globin-like 113449 px a.1.1.1 d1ux8a_ 1ux8 A: ^1 ^2 ^3 ^4 ^5
Five tab-delimited columns:
- sunid
- level: cl - class; cf - fold; sf - superfamily; fa - family; dm - protein; sp - species; px - domain
- sccs
- sid or "-"
- description
-
dir.cla.scop(e).txt - Full classification for each domain.
d1ux8a_ 1ux8 A: a.1.1.1 113449 cl=46456,cf=46457,sf=46458,fa=46459,dm=46460,sp=116748,px=113449 2gtld1 2gtl D:8-147 a.1.1.2 135658 cl=46456,cf=46457,sf=46458,fa=46463,dm=116758,sp=116759,px=135658 d1cph.1 1cph B:,A: g.1.1.1 43831 cl=56992,cf=56993,sf=56994,fa=56995,dm=56996,sp=56997,px=43831 ^1 ^2 ^3 ^4 ^5 ^6
Six tab-delimited columns:
- sid
- PDB ID
- description
- sccs
- sunid
- sunids of ancestor nodes in comma-delimited list, in the format "level=sunid". For level codes, see description for dir.des.scop(e).txt.
-
dir.hie.scop(e).txt - Children and parents for each node
0 - 46456,48724,51349,53931,56572,56835,56992,57942,58117,58231,58788 46457 46456 46458,46548 ^1 ^2 ^3
Three tab-delimited columns:
- sunid
- parent's sunid or "-"
- comma-separated list of children's sunids or "-"
-
dir.com.scop(e).txt - Human and machine-annotated comments
100067 ! complexed with cyn, hem, xe 63437 ! SQ Q10784 164742 ! automated match to d1s56b_ ! complexed with cyn, hem, na; mutant ^1 ^2 ^3
Two or more "!"-delimited columns:
- sunid
- first comment
- additional comments (one per column)
-
dir.inc.scop(e).txt - Human and machine-annotated structural heterogeneity in SCOPe clades. New in SCOPe 2.08-stable
a.137 nf a.177.1.1 as d1siga_ g.24.1.1 fr d2heyr2,d2heyt2,d2hevr2 ^1 ^2 ^3
Three tab-delimited columns:
- sccs identifier for the structurally heterogeneous clade
- two-letter code indicating the type of heterogeneity: md - multiple alternative domain divisions; re - repeat-based domains, with different numbers of repeat units in different domains; fr - fragment; as - additional (sub)domain(s); ie - additional insertion(s) or extension(s); ms - missing some secondary structures; ae - additional element(s); me - missing some element(s); nf - not a true fold; hf - heterogeneous fold; dn - different domains include different subsets of (sub)domains. For more details about each type, see the help section Identify structurally heterogeneous families to aid automated methods.
- sid(s) of domain(s) that belongs to this family and having the specified type of heterogeneity; if empty, then all domains belonging to the sccs have this type of heterogeneity.
-
dir.rep.scop(e).txt - Human-annotated repeat units, for domains recognized based on a smaller repeating unit. New in SCOPe 2.08-stable
a.7.12.1 d1vcta1 1vct A:9-107 a.7.14.1 d1wr0a1 1wr0 A:5-81 ^1 ^2 ^3
Three tab-delimited columns:
- sccs identifier for the family defined by a repeat
- sid of the domain in which the canonical repeat is defined
- description(s), the beginning and end of a single repeat unit within the domain, separated by commas if more than one representative unit is present
Backward compatibility with SCOP
- Applies to: SCOPe version 2.01 through current release
- Reference: 1
In order to facilitate use of SCOPe data by SCOP and Astral users, we provide SCOPe data in parseable files in the same formats as the SCOP and Astral databases. SCOPe uses the same stable identifiers (e.g., sunid, sid, sccs) as were used for prior releases of SCOP and Astral, and the same protocols previously used to assign new SCOP and Astral identifiers are also used in SCOPe. A history of all changes between consecutive releases of SCOP and SCOPe is available on the Stats & History page.
Stable releases and updates
- Applies to: SCOPe version 2.02 through current release
- Reference: 1
Like all previous SCOP and SCOPe releases, SCOPe 2.08 is "stable," meaning the data will not change until the next release (although we will continue to make improvements or bug fixes to the interface).
Our current infrastructure imports and classifies new PDB files on a weekly basis.
Starting with SCOPe 2.02, we are also producing periodic updates that add newly released PDB entries the classification (approximately monthly). These periodic updates add new entries to the current release, without affecting older domains. Sequences for the newly added chains and domains will not be included in the Astral subsets until the subsequent stable release. Periodic releases are named by appending the stable version number with the release date of the periodic release (e.g., "SCOPe version 2.02-2013-06-20"). Stable releases are explicitly labeled stable on the website and downloadable files, (e.g., "SCOPe version 2.02-stable") to avoid confusion in cases where different files representing either a stable release or a periodic update are available.
In addition to making new domains and PDB entries visible through the web interface, the periodic updates also include updated versions of:
- All SCOPe parseable files (available under the Downloads > Parseable Files & Software menu)
- The SCOPe MySQL database (available under the Downloads > Parseable Files & Software menu)
- The Astral RAF sequence maps and full sequence sets (available under the Downloads > Astral Sequences & Subsets menu)
Note that these periodic updates will not replace stable releases or the files we make available with each stable version. Because stable releases of SCOPe (and the Astral subsets) are commonly used for benchmarking, we plan to continue producing stable releases as well as periodic updates to those releases. The stable releases will include any changes (such as merging or splitting clades, moving clades, or changes and corrections to domain boundaries) that curators make to the classification.
The most recent periodic release corresponding to each version of SCOPe will be available on the website, in addition to the stable releases. Parseable files corresponding to previous periodic releases are available on request. It is also fairly trivial to reconstruct earlier periodic releases using subsequent ones from the same stable release, since the date when each node was added to SCOPe is shown on the Stats & History page) and also stored in the MySQL database. More recently added nodes never affect earlier ones (all changes are made in the subsequent stable release).
Manual curation in SCOPe
- Applies to: SCOPe version 2.04 through current release
- References: 2-4
In order to move toward our goal of complete coverage of the PDB, we have re-introduced manual curation of new Folds, Superfamilies, and Families in SCOPe 2.04. We examined and classified structures from the largest Pfam families that were not included in SCOP 1.75. In curating members of the 34 largest unrepresented Pfam families (each with at least 20 structures not previously in SCOP), we added 18 new Folds, 25 new Superfamilies, and 47 new Families to SCOPe 2.04. Aided by improvements to our automated curation method, we increased the fraction of classified PDB structures from 65% in SCOPe 2.03 to 68% in SCOPe 2.04.
Manual curation in SCOPe is performed by John-Marc Chandonia. Dr. Chandonia has reviewed Alexey Murzin's annotations since 2001 as part of the Astral build process, including dialog regarding manual classification errors. He has followed the same conservative principles as Dr. Murzin in deciding whether to annotate newly structurally characterized proteins as homologous to prior structures. We also invite collaborations with experts in particular superfamilies; if you would like to help us update part of the SCOPe hierarchy, please contact us at scope@compbio.berkeley.edu.
The figure below shows four examples of new Superfamilies and Families added in SCOPe 2.04:
- Macroglobulin: The complement system is an ancient system for immune response, pre-dating the immunoglobulins. Eight domains (red and pink) from mammalian C5 (3cu7) belong to different families in the new macroglobulin superfamily.
- Zn aminopeptidase insert domain: Several recently characterized proteases, including ERAP1 (2yd0), which is associated with chronic inflammatory disease [PubMed], contain an immunoglobulin-like domain not previously observed in proteases.
- DDB1-like: DDB1 is part of an important complex that regulates DNA repair, replication, and transcription [PubMed]. Three domains from 2b5l belong to this family: one is shown in red and the other two in pink. A novel C-terminal fold is also shown in blue.
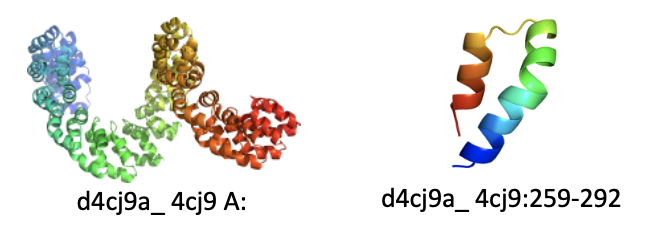
- PA14-like: The Anthrax Protective Antigen (APA) fold, classified in SCOP 1.75 as a single multi-domain fold, was split into N- and C-terminal portions because members of the PA14-like superfamily (e.g., d3abza3 shown in red) have recently been structurally characterized in a variety of different enzymes.
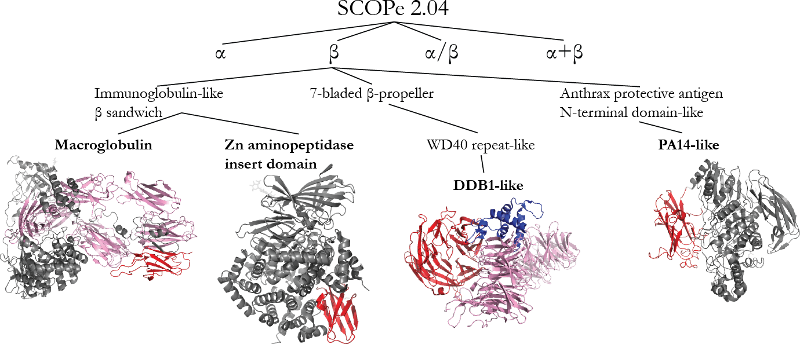

Automatic classification of PDB entries
- Applies to: SCOPe version 2.04 through current release
- References: 1,14
The vast majority of new protein structures solved represent a new experiment on a protein that was already structurally characterized (Chandonia and Brenner, 2006, available on the Help > References page). Our goal in implementing automation is classify structures that can be reliably classified in the existing SCOP hierarchy, without sacrificing the reliability of the database, which has been maintained through years of expert curation.
We introduced a new automated assigment protocol in SCOPe 2.01 (described here), which classified only single-domain chains, and extended it in SCOPe 2.03 (described here) to classify some multi-domain chains.
Starting with SCOPe 2.04, we modified some of the parameters in the 2.03 method in order to classify more chains. The list of changes from the previous version is:
- Removed the restriction on low-resolution, NMR, and ribosomal structures.
- Removed two-domain limit, permitting chains with any number of domains to be classified.
- Increased the number of residues by which we extend BLAST annotations to chain ends or gaps, from 10 to 15 residues.
- Removed the requirement that the BLAST hits used for the two domains in the chain not be to the same target SCOPe domain.
To ensure that these changes would not increase our error rate, we applied the same tests described in the Benchmarking section, below.
The updated procedure is described next. The figure below, taken
from [1], shows an
example application of the method:
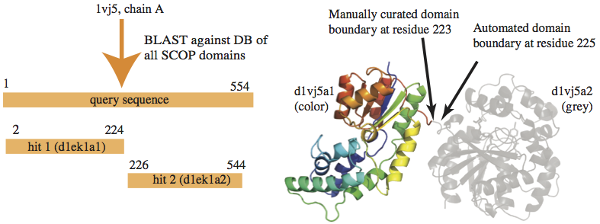

Description of Method:
We first create a BLAST database containing the SEQRES-based sequences for each domain in SCOPe. Then, for each newly released PDB chain, we BLAST its SEQRES sequence against the domain sequence database. BLAST performs local alignment, returning the start and end of the alignment (the hit) for the query sequence and the target sequence, as well as the E-value. We collect only the BLAST alignments where the E-value is at least as significant as 10-4 and the alignment covers most of the target domain (defined as missing at most 10 residues from each end). We group the alignments by the PDB chain to which the targets belong and rank the groups by the total number of residues covered by the hits on the query chain. We then use the top-ranking group of BLAST-based alignments to annotate domains in the query sequence. If the BLAST boundaries are outside the observed residues in the query chain, the assigned SCOPe domain is shortened to include only observed residues.
The boundaries are then extended by applying two methods. If the nearest PDB chain end or gap in ATOM records is within 15 residues of an alignment end, we extend the domain to include these residues. If a region between two alignment ends is 10 residues or shorter, we extend the two domain boundaries to fill the region. The purpose of extending the BLAST hit is to classify every observed residue (i.e., one with ATOM records) in the chain; the 15-residue limitation makes it very unlikely that the extension will include a new domain. Any longer gap between BLAST alignments or chain ends results in the chain being put on hold to await manual classification.
After making predictions for each chain, we apply some criteria to determine whether each domain prediction was high-confidence (i.e., sufficiently accurate to be included in SCOPe without further manual inspection). We first verify that each chain contains no domains that are homologous to genetic domains classified in SCOP (due to the difficulty of correctly automating predictions that include multiple PDB chains). In cases where an entire PDB chain was predicted to be a single SCOPe domain, we verify that the target domain also comprised its entire PDB chain. In cases where a PDB chain was divided into multiple domains, we verify that there are no regions in the ATOM records of the chain that are both longer than 10 residues and not assigned to any domain.
To fully classify domain predictions in the SCOPe hierarchy, we also had to assign levels below Superfamily. In our benchmarking, we found that the following rules could be used to reliably assign those levels:
- Family can be assigned if the BLAST percent identity to the previously classified domain is at least 55%, or if the gene annotation provided in the PDB file matched the gene annotation of exactly one family in the assigned Superfamily.
- Protein can be assigned if the BLAST percent identity to the previously classified domain is at least 99%, or if the gene annotation provided in the PDB file matched the gene annotation of exactly one protein in the assigned Family. This relatively high threshold is required because some paralogs in SCOP have ~98% identity to each other, and are classified as separate proteins.
- Species can not be reliably assigned based on sequence similarity, but can often be matched to a species in SCOP based on the species annotation in the PDB file.
In cases where the protein or family could not be reliably matched to an existing SCOP clade, we created a new group called "automated matches" at the Family or Protein level. These are not true families or proteins, because the domains within each one are not as similar to each other as members of manually curated families and proteins. For example, assignment to an "automated matches" family means the domain could reliably be assigned to the parent superfamily, but could not be automatically assigned to one of the existing families with 100% confidence. Manual curation or future improvements in automation may allow us to re-assign these domains with greater specificity. New SCOPe Species nodes were created if the PDB species annotation did not match any existing SCOP species.
To facilitate the mapping of SCOPe Species nodes to PDB species annotations, we attempted to make the descriptions of SCOPe species-level nodes more consistent, e.g., by always using the same common name for a species, and by updating NCBI taxonomy ids that have changed. This resulted in 1,520 species nodes being updated between SCOP 1.75 and SCOPe 2.01. The sunids for these nodes did NOT change, since each refers to the same species as before.
Benchmarking
- Applies to: SCOPe version 2.01 through current release
- Reference: 1
To validate the automatic classification method, we performed benchmarking against all SCOP releases with stable identifiers (i.e. releases 1.55-1.75). All PDB entries that were added between each pair of consecutive releases were automatically classified based on the earlier release and compared with the manually curated domains in the subsequent release. A predicted domain was considered identified and classified correctly if:
- Was classified in the same Superfamily as the manually curated domain.
- Had the same number of regions (i.e., consecutive parts of the sequence) as the manually curated domain.
- Had all region boundaries within 10 residues of the manually curated boundaries.
We have taken a very conservative approach to applying automated classification, in order to ensure that our methods are as accurate as manual curation, even though there are many proteins to which our methods cannot be applied. This means that our automated methods were tuned until they produced zero errors relative to manual curation. In fact, benchmarking revealed some previous errors in manual curation, which we corrected in SCOPe.
Annotating Families containing Structurally Heterogeneous Domains
- Applies to: SCOPe version 2.08 through current release
- Reference: 2
To facilitate automated methods developed or trained on the SCOPe database, we provide machine-parseable annotations of families containing structurally heterogeneous domains, in classes a to g.
Types of Structural Heterogeneity:
We first flagged potential heterogeneous families automatically, followed by manual review (see "Description of Method" for details). We manually assigned a heterogeneity type(s) to each domain. The heterogeneity types and their features are described below:
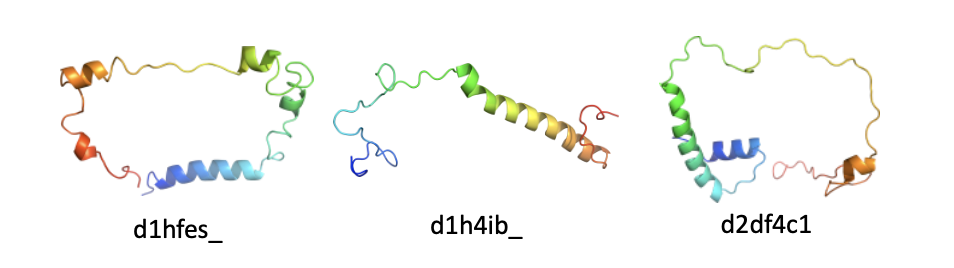
- Multiple alternative domain divisions: Applies to families that are inconsistently divided into domains. See figure below.

The three domains above belong to family b.11.1.1: Crystallins/Ca-binding development proteins. Most entries in this family are divided into domains containing 8 beta strands. However, d1bd7a_ remains undivided. Therefore, we label d1bd7a_ as having an alternative domain division. - Additional (sub)domain(s): Applies to domains that contains additional (sub)domain(s) that do not appear in the common domain. See figure below.
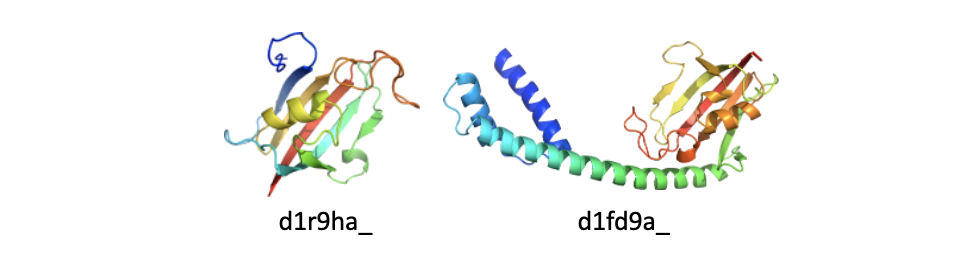
The two domains above belong to family d.26.1.1: FKBP immunophilin/proline isomerase. d1r9ha_(left) has the common family domain; d1fd9a_(right) shares the common family domain and contains an additional N-terminal all-alpha subdomain. - Additional insertion(s)/extension(s): Applies to domains that contain additional insertion(s) or extension(s) that are not arranged into (sub)domain(s). These can be additional secondary structures, significantly longer family-specific secondary structures, disordered loops, etc. See figure below.

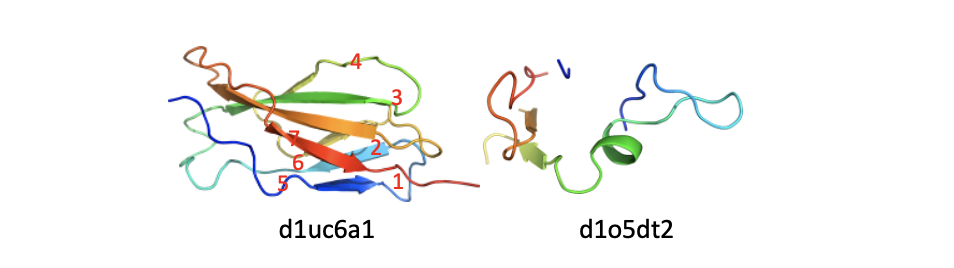
The two domains belong to family d.26.1.1: FKBP immunophilin/proline isomerase. d1r9ha_ (left) has the common family domain; d1jvwa_ (right) has the common family domain followed by an additional alpha helix at both termini. - Fragment: Applies to domains that are missing at least 1/3 of the common domain (either sequence-wise or structure-wise). See figure below.
The two domains belong to family b.1.2.1: Fibronectin type III. d1uc6a1 (left) is the common domain with 7 beta strands in 2 sheets; d1o5dt2 (right) is a fragment that is missing more than one third of the beta strands in the common domain. - Missing some secondary structure(s): Applies to domains that are missing secondary structures from the common domain (but still contain at least 2/3 of the structure, or these would be consider fragments). See figure below.

The two domains belong to family b.1.2.1: Fibronectin type III. d1uc6a1 (left) is the common domain with 7 beta strands in 2 sheets; d1bd2d2 (right) is missing one strand. - Additional element(s): Applies to domains that contain additional family-specific residue(s) or disulfide bond(s). See figure below.
- Missing element(s): Apply to a domain that is missing some secondary strcture elements or disulfide bonds. See figure below.
- Different number of (sub)domain(s) of a multi-domain family: Applies to all members of a family if the family is multi-domain by definition, but the members may contain any number of domain(s) or all domains. See figure below.
- Heterogenous fold: Applies to domains that adopt a different fold from the common domain but has similar sequence and number of secondary structures. This does not include domain swaps. See figure below.
- Not a true fold: Applies to all domains of a fold that was described by SCOP(e) curators as "not a true fold".
- Repeat and inconsistent: Applies to all domains of a family if the common domain is composed of different/variable number of small repeating units. (For all repeat families, whose common domain is composed of repeating units, we also provide a parseable file of a single unit per family.) See figure below.




The parseable files for these annotations are called "dir.inc" files, and users can find documentation of the format here.
Description of Method:
To locate potentially structurally heterogeneous domains, we assumed that if all domains in a family were structurally similar, the length of the domains would also be very similar. Thus, we collected domain length data and flagged families with significant length variation for manual review. Two different domain lengths were calculated: one based on PDB SEQRES records, the genetically encoded protein sequence; and one based on ATOM records, which were the parts of the protein that were experimentally observed (Brenner et al. 2000). For large families, we detected length variation by applying kernel density estimation to the SEQRES lengths and counting and comparing the resulting peaks. For small families, we detected length variation by applying kernel density estimation to the SEQRES lengths and counting and comparing the resulting peaks. For smaller families, we deployed a simpler method by calculating the ratio of the longest to shortest SEQRES length in the family. In addition, we also flagged domains and families if the SEQRES and ATOM sequence lengths were significantly different, or which contained human-readable metadata (clade names or comments) that suggested potential heterogeneity. We have also incorporated a version of this flagging system into our code for adding new, manually classified entries, in order to provide additional protection against manual errors such as typos.
This method is still not comprehensive. For example, it cannot detect a heterogeneous family in which all domains have similar lengths; it also does poorly in detecting domains with small additional or missing structures. However, it is effective in flagging major differences such as large insertions or deletions. We plan to improve our method by including structural comparison to help identify more heterogeneity, especially for heterogeneous folds.
Identification of Repeat Units
- Applies to: SCOPe version 2.08 through current release
- Reference: 2
Some protein domains in SCOPe consist of a number of smaller tandem
repeating units. The number of repeats may or may not be the same
between the domains in the same family. To facilitate automated
algorithms developed or trained on the SCOPe database, we provide
machine-parseable annotations of the extent of a single repeat
unit for all families of repeats in classes a to g. These
parseable files are called "dir.rep" files, and the format
is documented here.
Tandem repeats are often also annotated in other databases, such as Pfam.
Manual error correction of SCOP entries
- Applies to: SCOPe version 2.01 through current release
- Reference: 1
Over the course of benchmarking, we uncovered a number of errors in domains in SCOP. These were then manually corrected. We detected errors in 70 manually curated domains by running benchmarking and manually inspecting predicted domains that did not sufficiently match the manually annotated domains. These errors in domain boundaries in multi-domain chains were manually fixed in SCOPe 2.03. We also detected and fixed inconsistencies in 5,054 domains that had been predicted and classified with the SCOP 1.73 automated method. We review some of the types of errors detected.
The figure below, taken from
[1], shows four
examples of domains with incorrect boundaries that were corrected in
SCOPe releases:


- A previous automated method introduced in SCOP 1.73 used to predict domain d2p8qa1 had included approximately half the residues in the chain. This was inconsistent with all other manually curated entries in its species-level clade that included the entire chain. Compare d2p8qa1 in SCOP 1.75 to d2p8qa_ in SCOPe 2.01.
- A strand of beta sheet was included in the d1tqya2 domain by manual curation. Compare d2p8qa1 in SCOP 1.75 to d2p8qa1 in SCOPe 2.03.
- All of chain I from 1oyv had been placed into a single domain. Compare d1oyvi_ in SCOP 1.75 to d1oyv1 in SCOPe 2.03.
- The manually curated domain d1seja2 excluded the first helix in the chain. Compare d1seja2 in SCOP 1.75 and d1seja2 in SCOPe 2.03.
Plans for future website development
We are continuing to upgrade the website. Planned features include more sophisticated search functionality (including searches by sequence and by structure), use of AJAX to avoid full page reloads, and a SCOPe tree browser for more efficient display of results. We also plan to allow downloads of additional derived data (e.g., sequence sets and HMMs) for different clades in the tree.
Further help
The primary sources of documentation are the papers listed on the Help > References page.Please report bugs and usability issues to us at scope@compbio.berkeley.edu.